
Understanding the self-assembly of TCNQ on Cu(111): a combined study based on scanning tunnelling microscopy experiments and density functional theory simulations
Abstract
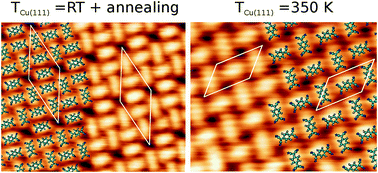
The structure of self-assembled monolayers of 7,7′,8,8′-tetracyano-p-quinodimethane (TCNQ) adsorbed on Cu(111) has been studied using a combination of scanning tunnelling microscopy (STM) experiments and density functional theory (DFT) calculations. We show that the polymorphism of the self-assembled molecular layer can be controlled by tuning of the experimental conditions under which the deposition is carried out. When the Cu(111) substrate is held above room temperature (TCu(111) = 350 K) during deposition, a structure is formed in which the two molecules in the unit cell are oriented one perpendicular to the other. Conversely, when the substrate is held at room temperature during deposition and slightly annealed afterwards, a more complex structure with five molecules per unit cell is formed. DFT calculations complement the experimental results by revealing that the building blocks of the two superstructures are two mutually orthogonal adsorption configurations of the molecule. The relative stability between the two observed polymorphs is reproduced by models of the two superstructures based on these two adsorption configurations.
 Please wait while we load your content...
Please wait while we load your content...

