
Reactive wetting properties of TiO2 nanoparticles predicted by ab initio molecular dynamics simulations†
Abstract
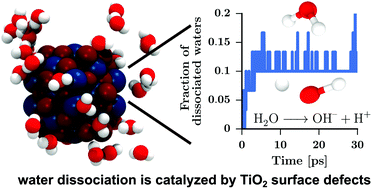
Small-sized wet TiO2 nanoparticles have been investigated by ab initio molecular dynamics simulations. Chemical and physical adsorption of water on the TiO2–water interface was studied as a function of water content, ranging from dry nanoparticles to wet nanoparticles with monolayer coverage of water. The surface reactivity was shown to be a concave function of water content and driven by surface defects. The local coordination number at the defect was identified as the key factor to decide whether water adsorption proceeds through dissociation or physisorption on the surface. A consistent picture of TiO2 nanoparticle wetting at the microscopic level emerges, which corroborates existing experimental data and gives further insight into the molecular mechanisms behind nanoparticle wetting. These calculations will facilitate the engineering of metal oxide nanoparticles with a controlled catalytic water activity.
 Please wait while we load your content...
Please wait while we load your content...

