
Crystal structure and modeled charge carrier mobility of benzobis(thiadiazole) derivatives†
Abstract
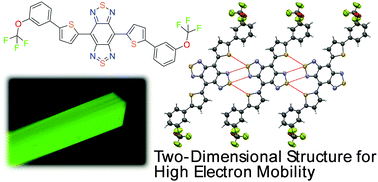
The crystal structures of benzobis(thiadiazole) (BBT)-based organic semiconductors with high electron mobilities were revealed using single-crystal X-ray analyses, followed by density functional theory and hopping modeling to estimate the charge carrier mobility levels. The crystal packings and intermolecular interactions were found to be similar regardless of trifluoromethyl and trifluoromethoxy groups for the corresponding para-substituted or meta-substituted derivatives. Although a compound with ortho-trifluoromethyl groups showed a molecule with a twisted conformation, the ortho-trifluoromethoxy derivative had nearly planar conformation. Since the ortho-trifluoromethyl groups prevent effective intermolecular interactions through short heteroatom contacts between BBT rings, molecules have a one-dimensional charge carrier transport path, resulting in lower overall charge carrier mobility in transistor devices. On the other hand, the ortho-trifluoromethoxy derivative exhibits a herringbone packing with large diffusion coefficients. The para and meta-derivatives showed an unusually large diffusion coefficient between the molecules located in the co-planar and the standard π–π stacking directions. The formation of a two-dimensional structure afforded high electron mobilities in actual organic thin-film transistor (OTFT) devices using these BBT-based materials.
 Please wait while we load your content...
Please wait while we load your content...

