
Versatile bonding and coordination modes of ditriazolylidene ligands in rhodium(iii) and iridium(iii) complexes†
Abstract
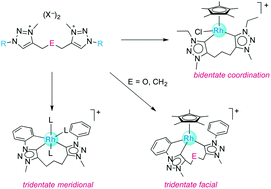
Metalation of novel ditriazolium salts containing a trimethylene (–CH2CH2CH2–) or dimethylether linker (–CH2OCH2–) was probed with different rhodium(III) and iridium(III) precursors. When using [MCp*Cl2]2, a transmetalation protocol via a triazolylidene silver intermediate was effective, while base-assisted metalation with MCl3via sequential deprotonation of the triazolium salt with KOtBu and addition of the metal precursor afforded homoleptic complexes. The N-substituent on the triazole heterocycle directed the metalation process and led to Ctrz,Ctrz,CPh-tridentate chelating ditriazolylidene complexes for N-phenyl substituents. With ethyl substituents, only Ctrz,Ctrz-bidentate complexes were formed, while metalation with mesityl substituents was unsuccessful, presumably due to steric constraints. Through modification of the reaction conditions for the metalation step, an intermediate species was isolated that contains a Ctrz,CPh-bidentate chelate en route to the formation of the tridentate ligand system. Accordingly, Cphenyl–H bond activation occurs prior to formation of the second metal–triazolylidene bond. Stability studies with a Ctrz,Ctrz,CPh-tridentate chelating ditriazolylidene iridium complex towards DCl showed deuterium incorporation at both N-phenyl groups and indicate that Cphenyl–H bond activation is reversible while the Ctrz–Ir bond is robust. The flexible linker between the two triazolylidene donor sites provides access to both facial and meridional coordination modes.
- This article is part of the themed collection: Reactions Facilitated by Ligand Design
 Please wait while we load your content...
Please wait while we load your content...

