
Anisotropic thermal motion in transition-metal carbonyls from experiments and ab initio theory†
Abstract
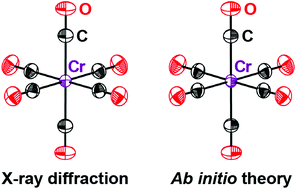
The thermal motion of atoms in crystals is quantified by anisotropic displacement parameters (ADPs). Here we show that dispersion-corrected periodic density-functional theory can be used to compute accurate ADPs for transition metal carbonyls, which serve as model systems for crystalline organometallic and coordination compounds.
 Please wait while we load your content...
Please wait while we load your content...

