
Thermodynamic and kinetic studies of LiNi0.5Co0.2Mn0.3O2 as a positive electrode material for Li-ion batteries using first principles†
Abstract
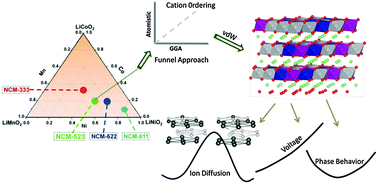
Ni-rich Li-based layered Ni, Co, and Mn (NCM) materials have shown tremendous promise in recent years as positive electrode materials for Li-ion batteries. This is evident as companies developing batteries for electrical vehicles are currently commercializing these materials. Despite the considerable research performed on LiNiαCoβMnγO2 systems, we do not yet have a complete atomic level understanding of these materials. In this work we study the cationic ordering, thermodynamics, and diffusion kinetics of LiNi0.5Co0.2Mn0.3O2 (NCM-523). Initially, we show that cationic ordering can be predicted employing cheap atomistic simulations, instead of using expensive first-principles methods. Subsequently, we investigate the electrochemical, thermodynamic and kinetic properties of NCM-523 using density functional theory (DFT). Our results demonstrate the importance of including dispersion corrections to standard first principles functionals in order to correctly predict the lattice parameters of layered cathode materials. We also demonstrate that a careful choice of computational protocol is essential to reproduce the experimental intercalation potential trends observed in the LiNi0.5Co0.2Mn0.3O2 electrodes. Analysis of the electronic structure confirms an active role of Ni in the electrochemical redox process. Moreover, we confirm the experimental finding that on complete delithiation, this material remains in an O3 phase, unlike LiCoO2 and NCM-333. Finally, we study various pathways for the Li-ion diffusion in NCM-523, and pinpoint the preferred diffusion channel based on first principles simulations. Interestingly, we observe that the Li diffusion barrier in NCM-523 is lower than that in LiCoO2.
- This article is part of the themed collection: 2016 most accessed PCCP articles
 Please wait while we load your content...
Please wait while we load your content...

