
Multiple binding modes of ibuprofen in human serum albumin identified by absolute binding free energy calculations†
Abstract
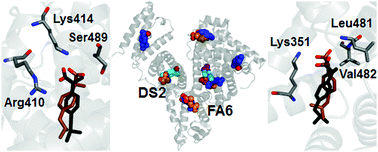
Human serum albumin possesses multiple binding sites and transports a wide range of ligands that include the anti-inflammatory drug ibuprofen. A complete map of the binding sites of ibuprofen in albumin is difficult to obtain in traditional experiments, because of the structural adaptability of this protein in accommodating small ligands. In this work, we provide a set of predictions covering the geometry, affinity of binding and protonation state for the pharmaceutically most active form (S-isomer) of ibuprofen to albumin, by using absolute binding free energy calculations in combination with classical molecular dynamics (MD) simulations and molecular docking. The most favorable binding modes correctly reproduce several experimentally identified binding locations, which include the two Sudlow's drug sites (DS2 and DS1) and the fatty acid binding sites 6 and 2 (FA6 and FA2). Previously unknown details of the binding conformations were revealed for some of them, and formerly undetected binding modes were found in other protein sites. The calculated binding affinities exhibit trends which seem to agree with the available experimental data, and drastically degrade when the ligand is modeled in a protonated (neutral) state, indicating that ibuprofen associates with albumin preferentially in its charged form. These findings provide a detailed description of the binding of ibuprofen, help to explain a wide range of results reported in the literature in the last decades, and demonstrate the possibility of using simulation methods to predict ligand binding to albumin.
 Please wait while we load your content...
Please wait while we load your content...

