
Prediction of proton affinities of organic molecules using the any-particle molecular-orbital second-order proton propagator approach†
Abstract
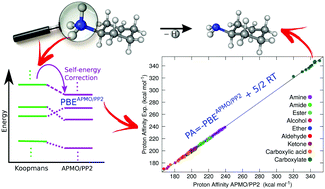
We assess the performance of the recently developed any-particle molecular-orbital second-order proton propagator (APMO/PP2) scheme [M. Díaz-Tinoco, J. Romero, J. V. Ortiz, A. Reyes and R. Flores-Moreno, J. Chem. Phys., 2013, 138, 194108] on the calculation of gas phase proton affinities (PAs) of a set of 150 organic molecules comprising several functional groups: amines, alcohols, aldehydes, amides, ketones, esters, ethers, carboxylic acids and carboxylate anions. APMO/PP2 PAs display an overall mean absolute error of 0.68 kcal mol−1 with respect to experimental data. These results suggest that the APMO/PP2 method is an alternative approach for the quantitative prediction of gas phase proton affinities. One novel feature of the method is that a PA can be obtained from a single calculation of the optimized protonated molecule.
 Please wait while we load your content...
Please wait while we load your content...

