
Ultrafast relaxation dynamics of electronically excited piperidine: ionization signatures of Rydberg/valence evolution
Abstract
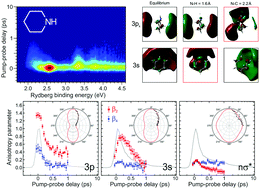
We have investigated the electronic relaxation dynamics of gas-phase piperidine (a secondary aliphatic amine) using time-resolved photoelectron imaging. Following 200 nm excitation, spectrally sharp and highly anisotropic photoelectron data reveal ultrafast (60 fs) internal conversion between the initially excited 3px Rydberg state and the lower-lying 3s Rydberg state, mediated by the evolution of nσ* valence character along the 3px N–C bond. This behaviour is in good agreement with previously reported findings for several tertiary aliphatic amines. In contrast to the these systems, however, much broader photoelectron signals exhibiting only very small angular anisotropy and two distinct decay timescales (180 fs and 1.7 ps) were also observed. As confirmed by our supporting calculations, this is attributable to nσ* valence character now evolving along the N–H stretching coordinate within the 3s Rydberg state as the molecule starts dissociating to yield H atom photoproducts in conjunction with ground state piperidinyl radicals. By analogy with systems such as ammonia and morpholine, we conclude this event may occur either promptly or, alternatively, via a “frustrated” process where the system repeatedly traverses the upper cone of a conical intersection with the ground state until the required region of phase space is sampled to facilitate non-adiabatic population transfer. Our findings reveal the role of several different nuclear coordinate motions in driving stepwise internal conversion across multiple potential energy surfaces and the distinct photoionization signatures that are associated with these processes.
 Please wait while we load your content...
Please wait while we load your content...

