
Planar B3S2H3− and B3S2H3 clusters with a five-membered B3S2 ring: boron–sulfur hydride analogues of cyclopentadiene†
Abstract
Boron clusters can serve as inorganic analogues of hydrocarbons or polycyclic aromatic hydrocarbons (PAHs). We present herein, based upon global searches and electronic structural calculations at the B3LYP and CCSD(T) levels, the global-minimum structures of two boron–sulfur hydride clusters: C2v B3S2H3− (1, 2B1) and C2v B3S2H3 (2, 1A1). Both species are perfectly planar and feature a five-membered B3S2 ring as the structural core, with three H atoms attached terminally to the B sites. Chemical bonding analysis shows that C2v B3S2H3− (1) has a delocalized 5π system within a heteroatomic B3S2 ring, analogous to the π bonding in cyclopentadiene, D5h C5H5. The corresponding closed-shell C2v B3S2H32− (3, 1A1) dianion is only a local minimum. At the single-point CCSD(T) level, it is 5.7 kcal mol−1 above the chain-like C1 (1A) open structure. This situation is in contrast to the cyclopentadienyl anion, C5H5−, a prototypical aromatic hydrocarbon with a π sextet. The C2v B3S2H3 (2) neutral cluster is readily obtained upon removal of one π electron from C2v B3S2H3− (1). The anion photoelectron spectrum of C2v B3S2H3− (1) and the infrared absorption spectrum of C2v B3S2H3 (2) are predicted. The C2v B3S2H3− (1) species can be stabilized in sandwich-type C2h [(B3S2H3)2Fe]2− and salt C2h [(B3S2H3)2Fe]Li2 complexes. An intriguing difference is observed between the pattern of π sextet in C2v B3S2H32− (3) dianion and that in cyclopentadienyl anion. The present work also sheds light on the mechanism of structural evolution in the B3S2H30/−/2− series with charge states.
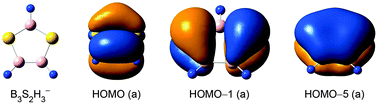
 Please wait while we load your content...
Please wait while we load your content...

