
A comparative study of AumRhn (4 ≤ m + n ≤ 6) clusters in the gas phase versus those deposited on (100) MgO†
Abstract
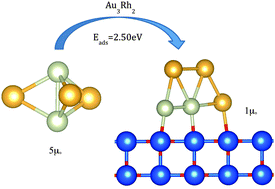
A comparative theoretical study has been performed of the gas phase and deposited AumRhn (4 ≤ m + n ≤ 6) clusters. The combined use of a genetic algorithm and Density Functional Theory (DFT) calculations allows us to explore the potential energy surface and, therefore, find efficiently and automatically the global minimum configuration for each composition. Our results show interesting effects on the geometries of the clusters on deposition. This occurs because the rhodium atoms (electronically) prefer to be in contact with the MgO surface, sometimes promoting planar clusters to become three-dimensional when deposited, and three-dimensional clusters in the gas phase to become two-dimensional. Together with the change in geometries, the magnetic moment is reduced from the gas phase, as the electrons rearrange themselves when the cluster interacts with the substrate.
 Please wait while we load your content...
Please wait while we load your content...

