
Structural phase transition in perovskite metal–formate frameworks: a Potts-type model with dipolar interactions
Abstract
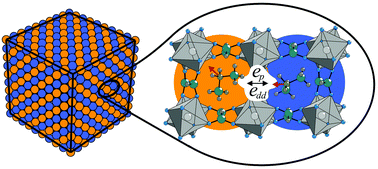
We propose a combined experimental and numerical study to describe an order–disorder structural phase transition in perovskite-based [(CH3)2NH2][M(HCOO)3] (M = Zn2+, Mn2+, Fe2+, Co2+ and Ni2+) dense metal–organic frameworks (MOFs). The three-fold degenerate orientation of the molecular (CH3)2NH2+ (DMA+) cation implies a selection of the statistical three-state model of the Potts type. It is constructed on a simple cubic lattice where each lattice point can be occupied by a DMA+ cation in one of the available states. In our model the main interaction is the nearest-neighbor Potts-type interaction, which effectively accounts for the H-bonding between DMA+ cations and M(HCOO)3− cages. The model is modified by accounting for the dipolar interactions which are evaluated for the real monoclinic lattice using density functional theory. We employ the Monte Carlo method to numerically study the model. The calculations are supplemented with the experimental measurements of electric polarization. The obtained results indicate that the three-state Potts model correctly describes the phase transition order in these MOFs, while dipolar interactions are necessary to obtain better agreement with the experimental polarization. We show that in our model with substantial dipolar interactions the ground state changes from uniform to the layers with alternating polarization directions.
 Please wait while we load your content...
Please wait while we load your content...

