
Rovibrational transitions of the methane–water dimer from intermolecular quantum dynamical computations†
Abstract
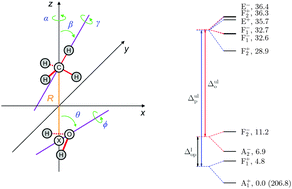
Rovibrational quantum nuclear motion computations, with J = 0, 1, and 2, are reported for the intermolecular degrees of freedom of the methane–water dimer, where J is the quantum number describing the overall rotation of the complex. The computations provide the first explanation of the far-infrared spectrum of this complex published in J. Chem. Phys., 1994, 100, 863. All experimentally reported rovibrational transitions, up to J = 2, can be assigned to transitions between the theoretically computed levels. The deviation of the experimental and computed rovibrational transitions is 0.5 cm−1 for the ortho and 2 cm−1 for the para species with a variance of 0.005 cm−1. In addition to a lower systematic error, the overall agreement of theory and experiment is also better for the ortho species (involving ortho-H2O). Most importantly, for this species all levels of the 24-fold tunneling splitting manifold corresponding to the zero-point vibration (ZPV) are involved in at least one experimentally reported transition. For the para species there are a few energy levels in the computed ZPV manifold that are not involved in the reported experimental transitions. Furthermore, computed energy levels are identified that correspond to the ZPV tunneling splitting manifold of the secondary minimum structure of the dimer, which presumably appear in rovibrational transitions in the same energy regime as the observed transitions, but have not been experimentally reported.
 Please wait while we load your content...
Please wait while we load your content...

