
Getting excited: challenges in quantum-classical studies of excitons in polymeric systems†
 *a
and
*a
and
Abstract
A combination of classical molecular dynamics (MM/MD) and quantum chemical calculations based on the density functional theory (DFT) was performed to describe the conformational properties of diphenylethyne (DPE), methylated-DPE and poly para phenylene ethynylene (PPE). DFT calculations were employed to improve and develop force field parameters for MM/MD simulations. Many-body Green's function theory within the GW approximation and the Bethe–Salpeter (GW-BSE) equation were utilized to describe the excited states of the systems. The reliability of the excitation energies based on the MM/MD conformations was examined and compared to the excitation energies from DFT conformations. The results show an overall agreement between the optical excitations based on MM/MD conformations and DFT conformations. This allows for the calculation of excitation energies based on MM/MD conformations.
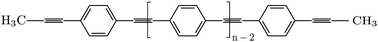
- This article is part of the themed collection: Insights from advanced methods in molecular dynamics
 Please wait while we load your content...
Please wait while we load your content...

