
The effect of the environment on the methyl transfer reaction mechanism between trimethylsulfonium and phenolate†
Abstract
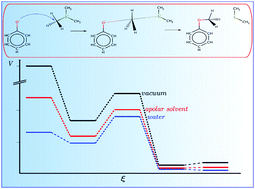
Methyl transfer reactions play an important role in biology and are catalyzed by various enzymes. Here, the influence of the molecular environment on the reaction mechanism was studied using advanced ab initio methods, implicit solvation models and QM/MM molecular dynamics simulations. Various conceptual DFT and electronic structure descriptors identified different processes along the reaction coordinate e.g. electron transfer. The results show that the polarity of the solvent increases the energy required for the electron transfer and that this spontaneous process is located in the transition state region identified by the (mean) reaction force analysis and takes place through the bonds which are broken and formed. The inclusion of entropic contributions and hydrogen bond interactions in QM/MM molecular dynamics simulations with a validated DFTB3 Hamiltonian yields activation barriers in good agreement with the experimental values in contrast to the values obtained using two implicit solvation models.
 Please wait while we load your content...
Please wait while we load your content...

