
Thermodynamic and redox properties of graphene oxides for lithium-ion battery applications: a first principles density functional theory modeling approach†
 *ac
and
*ac
and
Abstract
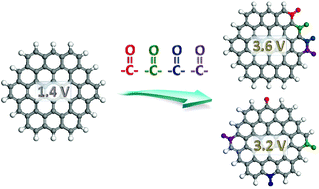
Understanding the thermodynamic stability and redox properties of oxygen functional groups on graphene is critical to systematically design stable graphene-based positive electrode materials with high potential for lithium-ion battery applications. In this work, we study the thermodynamic and redox properties of graphene functionalized with carbonyl and hydroxyl groups, and the evolution of these properties with the number, types and distribution of functional groups by employing the density functional theory method. It is found that the redox potential of the functionalized graphene is sensitive to the types, number, and distribution of oxygen functional groups. First, the carbonyl group induces higher redox potential than the hydroxyl group. Second, more carbonyl groups would result in higher redox potential. Lastly, the locally concentrated distribution of the carbonyl group is more beneficial to have higher redox potential compared to the uniformly dispersed distribution. In contrast, the distribution of the hydroxyl group does not affect the redox potential significantly. Thermodynamic investigation demonstrates that the incorporation of carbonyl groups at the edge of graphene is a promising strategy for designing thermodynamically stable positive electrode materials with high redox potentials.
 Please wait while we load your content...
Please wait while we load your content...

