
A molecular mechanical model for N-heterocyclic carbenes†
Abstract
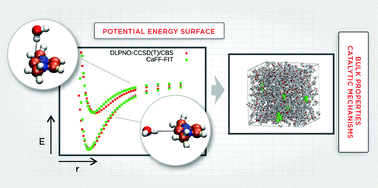
In this work we present a set of force fields for nine synthetically relevant and/or structurally interesting N-heterocyclic carbenes, including imidazol-, thiazol-, triazol-, imidazolidin-, and pyridine-ylidenes. The bonding parameters were calculated by using a series of geometry optimizations by ab initio methods. For fitting the non-bonding interactions, a water molecule was employed as a probe. The interaction energy between the carbene and the probe molecule was sampled along two coordinates for each carbene, representing the interaction through the lone pair, or the π system of the molecule. The corresponding reference interaction energies were obtained by CCSD(T)/CBS calculations. To describe the direction dependence of the intermolecular potential energy, an extra, massless Coulombic interaction site was included for all carbenes, which represents the lone pair of the divalent carbon atom. The resulting fitted carbene force field (CaFF) showed a robust behavior regarding probe molecule, as changing the molecular mechanical water model, or employing, instead, an OPLS methanol molecule did not introduce significant deviations in the potential energies. The obtained CaFF models are easy to merge with standard OPLS or AMBER force fields, therefore the molecular simulations of a large number of N-heterocyclic carbenes becomes available.
 Please wait while we load your content...
Please wait while we load your content...

