
On the physical origins of interaction-induced vibrational (hyper)polarizabilities†

Abstract
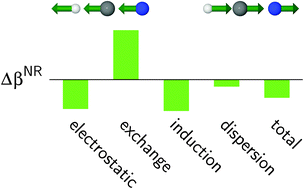
This paper presents the results of a pioneering exploration of the physical origins of vibrational contributions to the interaction-induced electric properties of molecular complexes. In order to analyze the excess nuclear relaxation (hyper)polarizabilities, a new scheme was proposed which relies on the computationally efficient Bishop–Hasan–Kirtman method for determining the nuclear relaxation contributions to electric properties. The extension presented herein is general and can be used with any interaction-energy partitioning method. As an example, in this study we employed the variational–perturbational interaction-energy decomposition scheme (at the MP2/aug-cc-pVQZ level) and the extended transition state method by employing three exchange–correlation functionals (BLYP, LC-BLYP, and LC-BLYP-dDsC) to study the excess properties of the HCN dimer. It was observed that the first-order electrostatic contribution to the excess nuclear relaxation polarizability cancels with the negative exchange repulsion term out to a large extent, resulting in a positive value of Δαnr due to the contributions from the delocalization and the dispersion terms. In the case of the excess nuclear relaxation first hyperpolarizability, the pattern of interaction contributions is very similar to that for Δαnr, both in terms of their sign as well as relative magnitude. Finally, our results show that the LC-BLYP and LC-BLYP-dDsC functionals, which yield smaller values of the orbital relaxation term than BLYP, are more successful in predicting excess properties.
 Please wait while we load your content...
Please wait while we load your content...

