
Configuration-averaged 4f orbitals in ab initio calculations of low-lying crystal field levels in lanthanide(iii) complexes
Abstract
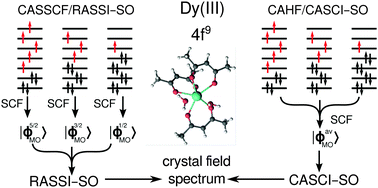
A successful and commonly used ab initio method for the calculation of crystal field levels and magnetic anisotropy of lanthanide complexes consists of spin-adapted state-averaged CASSCF calculations followed by state interaction with spin–orbit coupling (SI–SO). Based on two observations valid for Ln(III) complexes, namely: (i) CASSCF 4f orbitals are expected to change very little when optimized for different states belonging to the 4f electronic configuration, (ii) due to strong spin–orbit coupling the total spin is not a good quantum number, we show here via a straightforward analysis and direct calculation that the CASSCF/SI–SO method can be simplified to a single configuration-averaged HF calculation and one complete active space CI diagonalization, including spin–orbit coupling, on determinant basis. Besides its conceptual simplicity, this approach has the advantage that all spin states of the 4fn configuration are automatically included in the SO coupling, thereby overcoming one of the computational limitations of the existing CASSCF/SI–SO approach. As an example, we consider three isostructural complexes [Ln(acac)3(H2O)2], Ln = Dy3+, Ho3+, Er3+, and find that the proposed simplified method yields crystal field levels and magnetic g-tensors that are in very good agreement with those obtained with CASSCF/SI–SO.
 Please wait while we load your content...
Please wait while we load your content...

