
The equilibrium molecular structures of 2-deoxyribose and fructose by the semiexperimental mixed estimation method and coupled-cluster computations†
Abstract
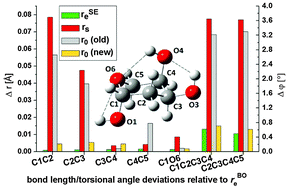
Fructose and deoxyribose (24 and 19 atoms, respectively) are too large for determining accurate equilibrium structures, either by high-level ab initio methods or by experiments alone. We show in this work that the semiexperimental (SE) mixed estimation (ME) method offers a valuable alternative for equilibrium structure determinations in moderate-sized molecules such as these monosaccharides or other biochemical building blocks. The SE/ME method proceeds by fitting experimental rotational data for a number of isotopologues, which have been corrected with theoretical vibration–rotation interaction parameters (αi), and predicate observations for the structure. The derived SE constants are later supplemented by carefully chosen structural parameters from medium level ab initio calculations, including those for hydrogen atoms. The combined data are then used in a weighted least-squares fit to determine an equilibrium structure (rSEe). We applied the ME method here to fructose and 2-deoxyribose and checked the accuracy of the calculations for 2-deoxyribose against the high level ab initio rBOe structure fully optimized at the CCSD(T) level. We show that the ME method allows determining a complete and reliable equilibrium structure for relatively large molecules, even when experimental rotational information includes a limited number of isotopologues. With a moderate computational cost the ME method could be applied to larger molecules, thereby improving the structural evidence for subtle orbital interactions such as the anomeric effect.
 Please wait while we load your content...
Please wait while we load your content...

