
Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: the prediction accuracy of sampling power and scoring power†
Abstract
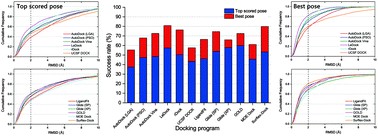
As one of the most popular computational approaches in modern structure-based drug design, molecular docking can be used not only to identify the correct conformation of a ligand within the target binding pocket but also to estimate the strength of the interaction between a target and a ligand. Nowadays, as a variety of docking programs are available for the scientific community, a comprehensive understanding of the advantages and limitations of each docking program is fundamentally important to conduct more reasonable docking studies and docking-based virtual screening. In the present study, based on an extensive dataset of 2002 protein–ligand complexes from the PDBbind database (version 2014), the performance of ten docking programs, including five commercial programs (LigandFit, Glide, GOLD, MOE Dock, and Surflex-Dock) and five academic programs (AutoDock, AutoDock Vina, LeDock, rDock, and UCSF DOCK), was systematically evaluated by examining the accuracies of binding pose prediction (sampling power) and binding affinity estimation (scoring power). Our results showed that GOLD and LeDock had the best sampling power (GOLD: 59.8% accuracy for the top scored poses; LeDock: 80.8% accuracy for the best poses) and AutoDock Vina had the best scoring power (rp/rs of 0.564/0.580 and 0.569/0.584 for the top scored poses and best poses), suggesting that the commercial programs did not show the expected better performance than the academic ones. Overall, the ligand binding poses could be identified in most cases by the evaluated docking programs but the ranks of the binding affinities for the entire dataset could not be well predicted by most docking programs. However, for some types of protein families, relatively high linear correlations between docking scores and experimental binding affinities could be achieved. To our knowledge, this study has been the most extensive evaluation of popular molecular docking programs in the last five years. It is expected that our work can offer useful information for the successful application of these docking tools to different requirements and targets.
- This article is part of the themed collection: Computational protein design and structure prediction: Celebrating the 2024 Nobel Prize in Chemistry
 Please wait while we load your content...
Please wait while we load your content...

