
First principles investigation of helium physisorption on an α-Al2O3(0001) surface
Abstract
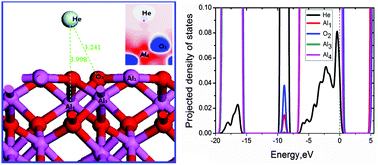
The interaction of helium with an α-Al2O3(0001) surface was studied by density functional theory (DFT), with consideration of the effects of He-coverage, surface defects, He-coadsorption and van der Waals interaction, respectively. Adsorption energies of helium atoms are very small as expected for a physisorbed state, varying from −20 to −5 meV, which is attributed to the small overlap between Al 3sp, O 2sp and He 1s states. A correlation is obtained for the adsorption energies and the He to nearest-neighbor Al atom distances on a clean (0001) surface. The He atom prefers to bound atop the Al site of the fourth atomic layer (Al4 hollow site), and the favorable site around an O-vacancy is atop the site of the O vacancy with less stability. The competition between O–He attraction and Al–He repulsion makes the He stable sites. As He-coverage on the surface increases, He atoms tend to form clusters, and coadsorption configuration is not solely determined by the most stable site but also by the He–He distance. The two co-adsorbed He atoms absorb on hollow sites Al4 and Al3, with a He–He distance of 2.767 Å. The OBS dispersion corrected DFT energies are 2.2–4.4 times larger than the non-corrected DFT values and He-surface distances are smaller. Finally, implications on H/He interaction within α-Al2O3 as a tritium permeation barrier are discussed.
 Please wait while we load your content...
Please wait while we load your content...

