
Effects of CO and CO2 on the desulfurization of H2S using a ZnO sorbent: a density functional theory study†
Abstract
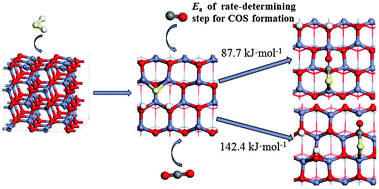
The density functional theory (DFT) method has been performed to study the effects of CO and CO2 on the desulfurization of H2S over a ZnO sorbent. It shows that COS is inevitably formed on the ZnO(10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) surface, which tends to be adsorbed onto the surface via a S–C bond binding with either a long or a short Zn–O bond. Potential energy profiles for the COS formation via reactions between H2S and CO, and H2S and CO2 on the ZnO(100) surface have been constructed. In the presence of CO, the dissociated active S of H2S reacting with CO leads to the formation of COS, and the activation energy of the rate-determining step is 87.7 kJ mol−1. When CO2 is present, the linear CO2 is first transferred to active CO2 in a triplet state, and then combines with active S to form COS with the highest energy barrier of 142.4 kJ mol−1. Rate constants at different temperatures show that the formation of COS via the reaction of CO and H2S is easier than that of CO2 and H2S over the ZnO surface.
0) surface, which tends to be adsorbed onto the surface via a S–C bond binding with either a long or a short Zn–O bond. Potential energy profiles for the COS formation via reactions between H2S and CO, and H2S and CO2 on the ZnO(100) surface have been constructed. In the presence of CO, the dissociated active S of H2S reacting with CO leads to the formation of COS, and the activation energy of the rate-determining step is 87.7 kJ mol−1. When CO2 is present, the linear CO2 is first transferred to active CO2 in a triplet state, and then combines with active S to form COS with the highest energy barrier of 142.4 kJ mol−1. Rate constants at different temperatures show that the formation of COS via the reaction of CO and H2S is easier than that of CO2 and H2S over the ZnO surface.
 Please wait while we load your content...
Please wait while we load your content...

