
Structural and mechanistic insight into substrate binding from the conformational dynamics in apo and substrate-bound DapE enzyme†
Abstract
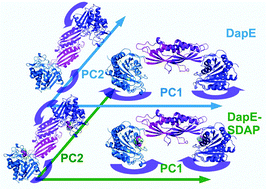
Conformational dynamics in large biomolecular systems is often associated with their physiological roles. The dynamics of a dimeric microbial enzyme, DapE, with great potential as an antibiotic target, has been studied employing long molecular dynamics simulations of the enzyme in apo form and in substrate bound complex form. The essential dynamics of the apo enzyme and the enzyme–substrate complex are extracted from the principal component analysis of the simulations of these two systems where the first two principal components are analyzed in detail. The essential motion of the enzyme in the substrate bound form exhibits a folding motion of its two catalytic domains over the two dimerization domains, which results in repulsion of water molecules away from the active site of the enzyme–substrate complex. This folding motion also leads to a stabilizing binding free energy of the substrate arising from the favorable interaction of the substrate and side chains of the enzyme. The dynamics in the enzyme–substrate complex results in stronger interaction between the catalytic and dimerization domains reflected by an increased number of inter-domain hydrogen bonds. The substrate, located in the catalytic domain of DapE, establishes contacts with the side chains of the dimerization domain of DapE by extended chains of hydrogen bonds, which emphasizes the role of the dimerization domain in substrate binding.
 Please wait while we load your content...
Please wait while we load your content...

