
Exceptional H2 sorption characteristics in a Mg2+-based metal–organic framework with small pores: insights from experimental and theoretical studies†
Abstract
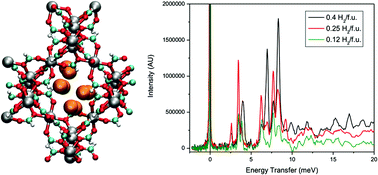
Experimental sorption measurements, inelastic neutron scattering (INS), and theoretical studies of H2 sorption were performed in α-[Mg3(O2CH)6], a metal–organic framework (MOF) that consists of a network of Mg2+ ions coordinated to formate ligands. The experimental H2 uptake at 77 K and 1.0 atm was observed to be 0.96 wt%, which is quite impressive for a Mg2+-based MOF that has a BET surface area of only 150 m2 g−1. Due to the presence of small pore sizes in the MOF, the isosteric heat of adsorption (Qst) value was observed to be reasonably high for a material with no open-metal sites (ca. 7.0 kJ mol−1). The INS spectra for H2 in α-[Mg3(O2CH)6] is very unusual for a porous material, as there exist several different peaks that occur below 10 meV. Simulations of H2 sorption in α-[Mg3(O2CH)6] revealed that the H2 molecules sorbed at three principal locations within the small pores of the framework. It was discovered through the simulations and two-dimensional quantum rotation calculations that different groups of peaks correspond to particular sorption sites in the material. However, for H2 sorbed at a specific site, it was observed that differences in the positions and angular orientations led to distinctions in the rotational tunnelling transitions; this led to a total of eight identified sites. An extremely high rotational barrier was calculated for H2 sorbed at the most favorable site in α-[Mg3(O2CH)6] (81.59 meV); this value is in close agreement to that determined using an empirical phenomenological model (75.71 meV). This rotational barrier for H2 exceeds those for various MOFs that contain open-metal sites and is currently the highest yet for a neutral MOF. This study highlights the synergy between experiment and theory to extract useful and important atomic level details on the remarkable sorption mechanism for H2 in a MOF with small pore sizes.
 Please wait while we load your content...
Please wait while we load your content...

