
Prediction of the Chapman–Jouguet chemical equilibrium state in a detonation wave from first principles based reactive molecular dynamics†
Abstract
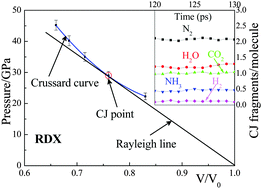
The combustion or detonation of reacting materials at high temperature and pressure can be characterized by the Chapman–Jouguet (CJ) state that describes the chemical equilibrium of the products at the end of the reaction zone of the detonation wave for sustained detonation. This provides the critical properties and product kinetics for input to macroscale continuum simulations of energetic materials. We propose the ReaxFF Reactive Dynamics to CJ point protocol (Rx2CJ) for predicting the CJ state parameters, providing the means to predict the performance of new materials prior to synthesis and characterization, allowing the simulation based design to be done in silico. Our Rx2CJ method is based on atomistic reactive molecular dynamics (RMD) using the QM-derived ReaxFF force field. We validate this method here by predicting the CJ point and detonation products for three typical energetic materials. We find good agreement between the predicted and experimental detonation velocities, indicating that this method can reliably predict the CJ state using modest levels of computation.
 Please wait while we load your content...
Please wait while we load your content...

