
Thermodynamic investigation of Ti doping in MgAl2O4 based on the first-principles method†
Abstract
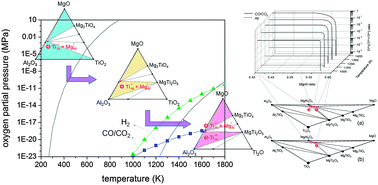
Ti doping of MgAl2O4 crystals is investigated using a theoretical thermodynamic approach. A number of types of Ti-doped MgAl2O4 are produced by combining possible Ti oxidation states (Ti2+, Ti3+, and Ti4+), doping sites (Al and Mg sites), and additional point defects (antisites and vacancies). Crystal models containing each doping type are prepared, and by treating them as independent phases, phase diagrams of the Mg–Al–Ti–O system are simulated based on first-principles calculations. This enables the study of stable doping types as a function of synthesis conditions and chemical compositions. Under air or H2 conditions, only the 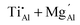 type is stable in the phase diagram, whereas
type is stable in the phase diagram, whereas 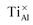 and
and 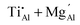 appear to be stable in a strongly reducing CO/CO2 atmosphere at high temperatures. A quantitative analysis model is established for calculating the fractions of certain doping types based on considerations of formation energy and mixing entropy, and the proportions of doping types are calculated. Changes in the Ti3+ : Ti4+ ratio are simulated, and the effects of temperature, oxygen partial pressure, Mg/Al ratio, and surface are examined. The results obtained show good agreement with XPS results and photoluminescence measurements. This study provides guidelines to the design and property tuning of Ti-doped MgAl2O4 phosphors for LED applications.
appear to be stable in a strongly reducing CO/CO2 atmosphere at high temperatures. A quantitative analysis model is established for calculating the fractions of certain doping types based on considerations of formation energy and mixing entropy, and the proportions of doping types are calculated. Changes in the Ti3+ : Ti4+ ratio are simulated, and the effects of temperature, oxygen partial pressure, Mg/Al ratio, and surface are examined. The results obtained show good agreement with XPS results and photoluminescence measurements. This study provides guidelines to the design and property tuning of Ti-doped MgAl2O4 phosphors for LED applications.
 Please wait while we load your content...
Please wait while we load your content...

