
A theoretical study on borenium ion affinities toward ammonia, formaldehyde and chloride anions†
Abstract
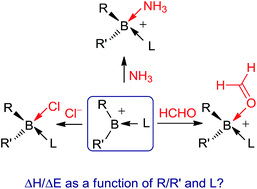
Various borenium ion affinities toward three ligands (L′ = NH3, HCHO and Cl−) have been evaluated by DFT calculations in the gas-phase and in solvent (CH2Cl2). The gas-phase results have been rationalized on the basis of quantitative decomposition of the total binding energy into contributions from electrostatic, orbital, dispersion and Pauli interactions, and energy needed to deform the interacting fragments from their optimal geometry to that they adopt in an adduct. Twenty six borenium cations, differing in the type of the two R/R′ substituents covalently bound to the boron atom and the neutral stabilizing ligand L, have been examined. With a few exceptions, the most important stabilizing interaction is electrostatic, more pronounced in the case of the charged ligand Cl−. Next come orbital interactions, involving the coordinate covalent bond formation, other charge transfer interactions between the cation and ligand, and polarization. Dispersion forces provide the smallest attraction, except in four complexes with long B–L′ distances. We present how substituent (R/R′)/ligand (L) variations affect binding enthalpies (ΔH)/energies (ΔE). Our results also show that the observed trend in the magnitudes of ΔHs/ΔEs represents an interplay of the above mentioned (de)stabilizing energies, and can be explained by consideration of the boron–ligand distance and all charge/orbital interactions, rather than partial ones involving boron and ligand L′. Under solvent conditions, the Cl− affinities are drastically reduced and made very similar to NH3 affinities, but still larger than HCHO affinities.
 Please wait while we load your content...
Please wait while we load your content...

