
Theoretical model estimation of guest diffusion in metal–organic frameworks (MOFs)
Abstract
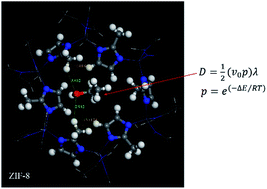
Characterizing molecule diffusion in nanoporous matrices is critical to understanding the novel chemical and physical properties of metal–organic frameworks (MOFs). In this paper, we developed a theoretical model to rapidly and accurately compute the diffusion rate of guest molecules in a zeolitic imidazolate framework-8 (ZIF-8). The ideal gas or equilibrium solution diffusion model was modified to contain the effect of host framework via introducing the possibility of guests passing through the framework gate. The only input in our model is the energy barrier of guests passing through the MOF's gate. Molecular dynamics (MD) methods were employed to gather the guest density profile, which then was used to deduce the energy barrier values. This produced reliable results that require a simulation time of 5 picoseconds, which is much shorter when using pure MD methods (in the nanosecond scale). Also, we used density functional theory (DFT) methods to obtain the energy profile of guests passing through gates, as this does not require specification of a force field for the MOF degrees of freedom. In the DFT calculation, we only considered one gate of MOFs each time; as this greatly reduced the computational cost. Based on the obtained energy barrier values we computed the diffusion rate of alkane and alcohol in ZIF-8 using our model, which was in good agreement with experimental test results and the calculation values from a standard MD model. Our model shows the advantage of obtaining accurate diffusion rates for guests in MOFs for a lower computational cost and shorter calculation time. Thus, our analytic model calculation is especially attractive for high-throughput computational screening of the dynamic performance of guests in a framework.
 Please wait while we load your content...
Please wait while we load your content...

