
Molecular dynamics simulations of self-assembled peptide amphiphile based cylindrical nanofibers†
Abstract
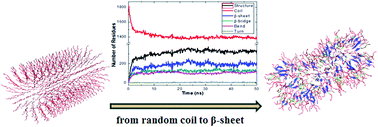
We carried out united-atom molecular dynamics simulations to understand the structural properties of peptide amphiphile (PA)-based cylindrical nanofibers and the factors that play a role in the “Self-Assembly” process on some specific nanofibers. In our simulations, we start from various cylindrical nanofiber structures with a different number of layers and a different number of PAs in each layer, based on previous experimental and theoretical results. We find that the 19-layered nanofiber, with 12 PAs at each layer, distributed radially and uniformly with alkyl chains in the center, is the most stable configuration with a diameter of 8.4 nm which is consistent with experimental results. The most dominant secondary structures formed in the fibers are random coils and β-sheets, respectively. We also find that hydrophobic interactions between the VVAG–VVAG moiety of the PA molecules and electrostatic interactions between D–Na+ and between E–R are responsible for the fiber's self-assembly properties. During the aggregation process, first dimers, then trimers are formed.
 Please wait while we load your content...
Please wait while we load your content...

