
Accurate ab initio calculations of O–H⋯O and O–H⋯−O proton chemical shifts: towards elucidation of the nature of the hydrogen bond and prediction of hydrogen bond distances†
Abstract
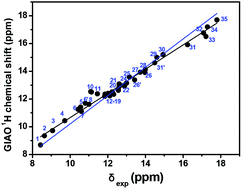
The inability to determine precisely the location of labile protons in X-ray molecular structures has been a key barrier to progress in many areas of molecular sciences. We report an approach for predicting hydrogen bond distances beyond the limits of X-ray crystallography based on accurate ab initio calculations of O–H⋯O proton chemical shifts, using a combination of DFT and contactor-like polarizable continuum model (PCM). Very good linear correlation between experimental and computed (at the GIAO/B3LYP/6-311++G(2d,p) level of theory) chemical shifts were obtained with a large set of 43 compounds in CHCl3 exhibiting intramolecular O–H⋯O and intermolecular and intramolecular ionic O–H⋯−O hydrogen bonds. The calculated OH chemical shifts exhibit a strong linear dependence on the computed (O)H⋯O hydrogen bond length, in the region of 1.24 to 1.85 Å, of −19.8 ppm Å−1 and −20.49 ppm Å−1 with optimization of the structures at the M06-2X/6-31+G(d) and B3LYP/6-31+G(d) level of theory, respectively. A Natural Bond Orbitals (NBO) analysis demonstrates a very good linear correlation between the calculated 1H chemical shifts and (i) the second-order perturbation stabilization energies, corresponding to charge transfer between the oxygen lone pairs and σ*OH antibonding orbital and (ii) Wiberg bond order of the O–H⋯O and O–H⋯−O hydrogen bond. Accurate ab initio calculations of O–H⋯O and O–H⋯−O 1H chemical shifts can provide improved structural and electronic description of hydrogen bonding and a highly accurate measure of distances of short and strong hydrogen bonds.
 Please wait while we load your content...
Please wait while we load your content...

