
Watson–Crick base pairing, electronic and photophysical properties of triazole modified adenine analogues: a computational study†
Abstract
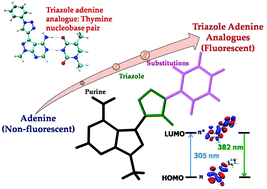
We employ first-principles Density Functional Theory (DFT) and time-dependent DFT (TDDFT) to elucidate structural, electronic and optical properties of a few recently reported triazole adenine nucleobase analogues. The results are compared against the findings obtained for both the natural adenine nucleobase and available experimental data. The optical absorption of these adenine analogues is calculated both in the gas-phase and in the solvent (methanol) using the Polarized Continuum Model (PCM). We find that all the analogues show a red-shifted absorption profile as compared to adenine. Our simulated emission spectra in the solvent compared fairly well with experimentally observed results. We investigate base paring ability of these adenine analogues with thymine. The calculations of the intrinsic stability of these base pairs ascertain that all the adenine analogues form a hydrogen bonded Watson–Crick base pair with similar H-bonding energy to that obtained for a natural adenine–thymine base pair. In our study, we provide a microscopic origin of the low-energy absorption and emission peaks, observed experimentally.
 Please wait while we load your content...
Please wait while we load your content...

