
Helical arylamide foldamers: structure prediction by molecular dynamics simulations†
Abstract
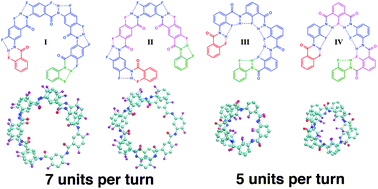
We present a molecular dynamics (MD) study on a series of helical arylamide oligomers with systematically varying building blocks and linkage types. This study showcases a computational approach for the prediction of secondary structure properties of arylamide foldamers and their solution dynamics. We demonstrate that the conformational characteristics of foldamers, such as the number of units per turn, helical pitch, and pore diameter, can be predicted by MD simulations of small oligomers significantly shorter than the foldamers in question. Importantly, the curvature angle, the key geometrical parameter in helical arylamide structures, can be accurately determined by MD simulation of tetramers, entities with often less than one helical turn. The curvature angle is found to be a local property associated with one single residue/unit, which enables highly accurate predictive power for designing oligomers with various scaffolds and sizes. In addition, MD simulations with the improved force field parameters capture solvent effects in terms of both protic solvent competition with intramolecular H-bonds and solvophobic effects. The computational approach can provide useful insight into dynamical, mechanistic and functional properties of the arylamide oligomer class, which will facilitate rational design of foldamers.
- This article is part of the themed collection: Foldamer Chemistry
 Please wait while we load your content...
Please wait while we load your content...

