
Decomposition of RNA methylome reveals co-methylation patterns induced by latent enzymatic regulators of the epitranscriptome†
Abstract
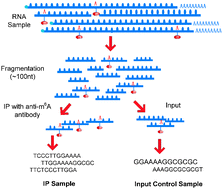
Biochemical modifications to mRNA, especially N6-methyladenosine (m6A) and 5-methylcytosine (m5C), have been recently shown to be associated with crucial biological functions. Despite the intriguing advancements, little is known so far about the dynamic landscape of RNA methylome across different cell types and how the epitranscriptome is regulated at the system level by enzymes, i.e., RNA methyltransferases and demethylases. To investigate this issue, a meta-analysis of m6A MeRIP-Seq datasets collected from 10 different experimental conditions (cell type/tissue or treatment) is performed, and the combinatorial epitranscriptome, which consists of 42 758 m6A sites, is extracted and divided into 3 clusters, in which the methylation sites are likely to be hyper- or hypo-methylated simultaneously (or co-methylated), indicating the sharing of a common methylation regulator. Four different clustering approaches are used, including K-means, hierarchical clustering (HC), Bayesian factor regression model (BFRM) and nonnegative matrix factorization (NMF) to unveil the co-methylation patterns. To validate whether the patterns are corresponding to enzymatic regulators, i.e., RNA methyltransferases or demethylases, the target sites of a known m6A regulator, fat mass and obesity-associated protein (FTO), are identified from an independent mouse MeRIP-Seq dataset and lifted to human. Our study shows that 3 out of the 4 clustering approaches used can successfully identify a group of methylation sites overlapping with FTO target sites at a significance level of 0.05 (after multiple hypothesis adjustment), among which, the result of NMF is the most significant (p-value 2.81 × 10−06). We defined a new approach evaluating the consistency between two clustering results which shows that clustering results of different methods are highly correlated strongly indicating the existence of co-methylation patterns. Consistent with recent studies, a number of cancer and neuronal disease-related bimolecular functions are enriched in the identified clusters, which are biological functions that can be regulated at the epitranscriptional level, indicating the pharmaceutical prospect of RNA N6-methyladenosine-related studies. This result successfully reveals the linkage between the global RNA co-methylation patterns embedded in the epitranscriptomic data under multiple experimental conditions and the latent enzymatic regulators, suggesting a promising direction towards a more comprehensive understanding of the epitranscriptome.
 Please wait while we load your content...
Please wait while we load your content...