
Tuning the ferroelectric polarization in AA′MnWO6 double perovskites through A cation substitution†
Abstract
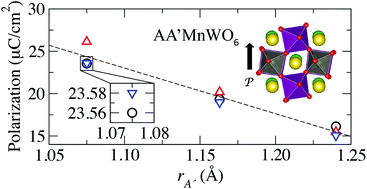
Recent experimental and theoretical work has shown that the double perovskite NaLaMnWO6 exhibits antiferromagnetic ordering owing to the Mn d states, and computational studies further predict it to exhibit a spontaneous electric polarization due to an improper mechanism for ferroelectricity [King et al., Phys. Rev. B: Condens. Matter, 2009, 79, 224428; Fukushima et al., Phys. Chem. Chem. Phys., 2011, 13, 12186], which make it a candidate multiferroic material. Using first-principles density functional calculations, we investigate nine isostructural and isovalent AA′MnWO6 double perovskites (A = Na, K, and Rb; A′ = La, Nd, and Y) with the aim of articulating crystal-chemistry guidelines describing how to enhance the magnitude of the electric polarization through chemical substitution of the A-site while retaining long-range magnetic order. We find that the electric polarization can be enhanced by up to 150% in compounds which maximize the difference in the ionic size of the A and A′ cations. By examining the tolerance factors, bond valences, and structural distortions (described by symmetry-adapted modes) of the nine compounds, we identify the atomic scale features that are strongly correlated with the ionic and electronic contributions to the electric polarization. We also find that each compound exhibits a purely electronic remnant polarization, even in the absence of a displacive polar mode. The analysis and design strategies presented here can be further extended to additional members of this family (B = Fe, Co, etc.), and the improper ferroelectric nature of the mechanism allows for the decoupling of magnetic and ferroelectric properties and the targeted design of novel multiferroics.
- This article is part of the themed collection: Perovskites
 Please wait while we load your content...
Please wait while we load your content...

