
Theoretical studies of energetics and binding isotope effects of binding a triazole-based inhibitor to HIV-1 reverse transcriptase†
Abstract
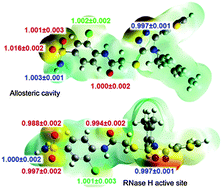
Understanding of protein-ligand interactions is crucial for rational drug design. Binding isotope effects, BIEs, can provide intimate details of specific interactions between individual atoms of an inhibitor and the binding pocket. We have applied multi-scale QM/MM simulations to evaluate binding energetics of a novel triazole-based non-nucleoside inhibitor of HIV-1 reverse transcriptase and to calculate associated BIEs. The binding sites can be distinguished based on the 18O-BIE.
 Please wait while we load your content...
Please wait while we load your content...

