
Dynamic and static behavior of hydrogen bonds of the X–H⋯π type (X = F, Cl, Br, I, RO and RR′N; R, R′ = H or Me) in the benzene π-system, elucidated by QTAIM dual functional analysis†
Abstract
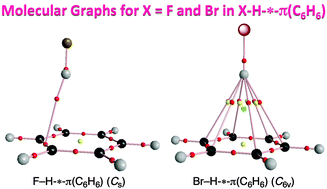
Dynamic and static behavior of the X–H-*-π interactions in X–H-*-π(C6H6) (X = F, Cl, Br, I, HO, MeO, H2N, MeHN and Me2N) is elucidated by QTAIM-DFA (QTAIM dual functional analysis), which we proposed recently, as the first step to clarify various types of X–H-*-π interactions. The asterisk * emphasizes the existence of the bond critical point (BCP) on the interaction in question. Total electron energy densities (Hb(rc)) are plotted versus Hb(rc) − Vb(rc)/2 [=(ℏ2/8m)∇2ρb(rc)] at BCPs in QTAIM-DFA, where Vb(rc) are potential energy densities at BCPs. In our treatment, data for the perturbed structures around the fully optimized ones are employed, in addition to those for the fully optimized structures. Data from the fully optimized structures are analyzed by the polar coordinate (R, θ) representation. Each plot for an interaction, containing data from the perturbed structures, shows a specific curve, which provides important information. The plot is expressed by (θp, κp): θp corresponds to the tangent line of the plot and κp is the curvature. θ and θp are measured from the y-axis and the y-direction, respectively. While (R, θ) correspond to the static nature, (θp, κp) represent the dynamic nature of interactions. The nature of the X–H-*-π(C6H6) interactions is well specified by (R, θ) and (θp, κp). All interactions, examined in this work, are classified by the pure closed shell interactions and predicted to have the van der Waals nature.
 Please wait while we load your content...
Please wait while we load your content...

