
Structure, solvent, and relativistic effects on the NMR chemical shifts in square-planar transition-metal complexes: assessment of DFT approaches†
 *ae
*ae
Abstract
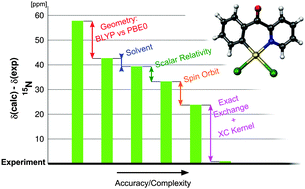
The role of various factors (structure, solvent, and relativistic treatment) was evaluated for square-planar 4d and 5d transition-metal complexes. The DFT method for calculating the structures was calibrated using a cluster approach and compared to X-ray geometries, with the PBE0 functional (def2-TZVPP basis set) providing the best results, followed closely by the hybrid TPSSH and the MN12SX functionals. Calculations of the NMR chemical shifts using the two-component (2c, Zeroth-Order Regular Approximation as implemented in the ADF package) and four-component (4c, Dirac–Coulomb as implemented in the ReSpect code) relativistic approaches were performed to analyze and demonstrate the importance of solvent corrections (2c) as well as a proper treatment of relativistic effects (4c). The importance of increased exact-exchange admixture in the functional (here PBE0) for reproducing the experimental data using the current implementation of the 2c approach is partly rationalized as a compensation for the missing exchange–correlation response kernel. The kernel contribution was identified to be about 15–20% of the spin–orbit-induced NMR chemical shift, ΔδSO, which roughly corresponds to an increase in ΔδSO introduced by the artificially increased exact-exchange admixture in the functional. Finally, the role of individual effects (geometry, solvent, relativity) in the NMR chemical shift is discussed in selected complexes. Although a fully relativistic DFT approach is still awaiting the implementation of GIAOs for hybrid functionals and an implicit solvent model, it nevertheless provides reliable NMR chemical shift data at an affordable computational cost. It is expected to outperform the 2c approach, in particular for the calculation of NMR parameters in heavy-element compounds.
 Please wait while we load your content...
Please wait while we load your content...

