
Hydrogen binding energies and electronic structure of Ni–Pd particles: a clue to their special catalytic properties†
Abstract
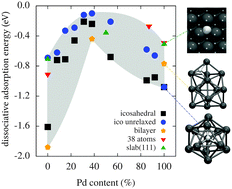
We investigate the electronic structure of nickel–palladium systems with first-principles density functional theory (DFT). Our study is motivated by the experimental observation that nickel–palladium nanoalloys containing approximately equal amounts of nickel and palladium show a higher catalytic activity in hydrogenation reactions than pure particles of either metal. Our calculations show that the energy with which hydrogen is bound to the metal surface, a decisive factor in catalytic activity, strongly depends on the nickel–palladium ratio. The weakest binding and Gibbs free energies of hydrogen adsorption close to zero are found for systems with roughly equal amounts of nickel and palladium. While for extended Ni–Pd surfaces this observation can be explained by the well-established d-band model, for Ni–Pd clusters analysis of the electronic density of states shows a complex interplay of s- and d-orbital contributions. Studying extended surfaces further reveals that the formation of nanostructures on the surface can influence hydrogen adsorption pronouncedly. We discuss the important role that the exchange–correlation functional approximation plays in DFT calculations for nickel–palladium systems.
 Please wait while we load your content...
Please wait while we load your content...

