
Thermodynamic properties of neutral and charged oxygen vacancies in BaZrO3 based on first principles phonon calculations
Abstract
The structural, electronic and thermodynamic properties of neutral 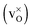 and positively doubly charged
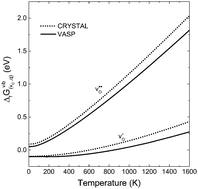
and positively doubly charged 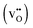 oxygen vacancies in BaZrO3 are addressed by first principles phonon calculations. The calculations are performed using two complementary first principles approaches and functionals; the linear combination of atomic orbitals (LCAO) within the hybrid Hartree–Fock and density functional theory formalism (HF-DFT), and the projector augmented plane wave approach (PAW) within DFT. Phonons are shown to contribute significantly to the formation energy of the charged oxygen vacancy at high temperatures (∼1 eV at 1000 K), due to both its large distortion of the local structure, and its large negative formation volume. For the neutral vacancy, the resulting lattice distortions, and thus the contributions from phonons to the free formation energy, are significantly smaller. As a result, phonons affect the relative stability of the two defects at finite temperatures and the charge transition level for oxygen vacancies (+2/0) changes from 0.42 to 0.83 eV below the conduction band bottom from 0 K to 1000 K.
oxygen vacancies in BaZrO3 are addressed by first principles phonon calculations. The calculations are performed using two complementary first principles approaches and functionals; the linear combination of atomic orbitals (LCAO) within the hybrid Hartree–Fock and density functional theory formalism (HF-DFT), and the projector augmented plane wave approach (PAW) within DFT. Phonons are shown to contribute significantly to the formation energy of the charged oxygen vacancy at high temperatures (∼1 eV at 1000 K), due to both its large distortion of the local structure, and its large negative formation volume. For the neutral vacancy, the resulting lattice distortions, and thus the contributions from phonons to the free formation energy, are significantly smaller. As a result, phonons affect the relative stability of the two defects at finite temperatures and the charge transition level for oxygen vacancies (+2/0) changes from 0.42 to 0.83 eV below the conduction band bottom from 0 K to 1000 K.
 Please wait while we load your content...
Please wait while we load your content...

