
Probing the π → π* photoisomerization mechanism of cis-azobenzene by multi-state ab initio on-the-fly trajectory dynamics simulation†
Abstract
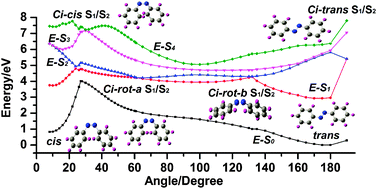
Based on a newly developed algorithm to compute global nonadiabatic switching probability by using only electronic adiabatic potential energy surfaces and gradients, we performed on-the-fly, trajectory-surface hopping simulations at the 5SA-CASSCF(6,6)/6-31G quantum level to probe the π → π* photoisomerization mechanism of the azobenzene within four singlet low-lying electronic states (S0, S1, S2, and S3) coupled with a complicated conical intersection network. We found that four conical intersections between the S1 and S2 states (one is near the cis-isomer region, another near the trans-isomer region, and two others between cis and trans) play the most important roles for understanding the photoisomerization mechanism of azobenzene upon S2 and S3 ππ* excitation. We studied six cases to demonstrate the photoisomerization mechanism in detail by choosing eight (six) typical reactive (nonreactive) trajectories, namely, two-step fast–fast processes having lifetimes of several tenths to one hundred femtoseconds and two-step, fast–slow and slow–slow processes having lifetimes of several hundred to one thousand femtoseconds. We found for the first time from simulation that once a trajectory visits the conical intersection near the trans-isomer after ππ* excitation, it could rapidly go through the inversion pathway to trans-azobenzene, and confirms the most recent experimental observations. We performed 536 sampling trajectories (336 from S2 and 200 from S3), initially starting from the Franck–Condon region of cis-azobenzene, and obtained a total reactive quantum yield of 0.3–0.45 in very good agreement with recent experimental results of 0.24–0.50. Moreover, the current method can estimate overall nonadiabatic transition probability for each sampling trajectory from beginning to end. This can greatly accelerate convergence of nonadiabatic molecular dynamic simulation, and, for instance, results in a quantum yield of 0.53 estimated from only eight typical reactive trajectories.
 Please wait while we load your content...
Please wait while we load your content...

