
To π or not to π – how does methanol dock onto anisole?†
 a
Martin A.
Suhm
*a
a
Martin A.
Suhm
*a
Abstract
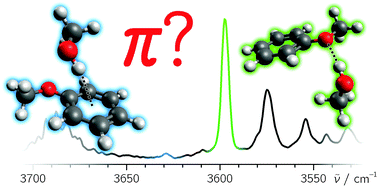
Anisole offers two similarly attractive hydrogen bond acceptor sites to an incoming hydrogen bond donor: its oxygen atom and its delocalized π electron system. Electronic structure calculations up to the CCSD(T)/AVTZ level suggest an isoenergetic situation for methanol after harmonic zero point energy correction, within less than 1 kJ mol−1. Linear infrared absorption spectroscopy in the OH stretching fundamental range applied to a cold supersonic jet expansion of anisole and methanol in helium shows that the oxygen binding site is preferred, with about 20 times less π-bonded than O-bonded dimers despite the non-equilibrium collisional environment. Accidental band overlap is ruled out by OH overtone and OD stretching spectroscopy. Furthermore, the diagonal anharmonicity constant of the OH stretching mode is derived from experiment and reaches 80% of the monomer distortion found in the methanol dimer, as expected for a weaker hydrogen bond to the aromatically substituted oxygen. To reconcile these experimental findings with ab initio theory, accurate nuclear and electronic structure calculations involving AVQZ basis sets are required. Dispersion-corrected double-hybrid density functional theory provides a less expensive successful structural approach.
 Please wait while we load your content...
Please wait while we load your content...

