
Electronic structure and conformational flexibility of d-cycloserine†
Abstract
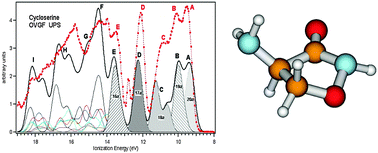
The first comprehensive investigation of the effect of conformational flexibility of gaseous D-cycloserine on the valence and core electronic structures is reported here. The seven most stable conformers among the twelve structures calculated at the MP2/6-311++G** level of theory were assumed to properly describe the properties of the investigated compound. Taking into account the contribution of these isomers, the valence photoelectron spectrum (UPS) was simulated by the Outer Valence Green' s Function (OVGF) method. A different sensitivity towards the conformational flexibility of the outermost photoelectron bands was exhibited in the simulated spectrum. The comparison of the theoretical UPS with the experimental one allowed a detailed assignment of the outermost valence spectral region. The composition and bonding properties of the relevant MOs of the most stable conformers were analyzed in terms of leading Natural Bond Orbital (NBO) contributions to the HF/6-311++G** canonical MOs. The C1s, N1s, and O1s photoelectron spectra (XPS) were theoretically simulated by calculating the vertical Ionization Energies (IEs) of the relevant conformers using the ΔSCF approach. The different IE chemical shift spread of the XPS components associated with various conformers, which is expected to affect the experimental spectra, could be evaluated by simulated XPS, thus providing a new insight into the core electronic structure. The comparison of the theoretical results with the experimental ones unraveled that the atomic XPS components are not mixed by conformational flexibility of D-cycloserine, and that the specific vibronic structure of different spectral components should play a crucial role in determining different relative intensities and band shapes observed in the experiment.
- This article is part of the themed collection: Optical spectroscopy coupled with mass spectrometry methods
 Please wait while we load your content...
Please wait while we load your content...

