
Predicting the degree of aromaticity of novel carbaporphyrinoids†
Abstract
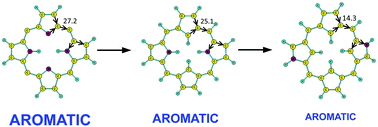
Magnetically induced current densities have been calculated for dioxaporphyrin, dithiaporphyrin, true carbaporphyrins, and N-confused porphyrins using the gauge-including magnetically induced current (GIMIC) method. The current-strength susceptibilities (current strengths) have been obtained by numerically integrating the current flow passing selected chemical bonds. The current strength calculations yield very detailed information about the electron delocalization pathways of the molecules. The strength of the ring-current that circles around the porphyrinoid macroring is used to estimate the degree of molecular aromaticity. The studied porphyrinoid structures have been obtained by replacing the NH and N groups of porphin with formally isoelectronic moieties such as O, S, CH and CH2. Replacing an NH moiety of trans-porphin with isoelectronic O and S does not significantly change the current strengths and pathways, whereas substitution of N with an isoelectronic CH group leads to significant changes in the current pathway and current strengths. CH2 groups cut the flow of diatropic currents, whereas in strongly antiaromatic molecules a significant fraction of the paratropic ring-current is able to pass the sp3 hybridized inner carbons. N-confused porphyrinoids sustain a ring current whose strength is about half the ring-current strength of porphin with the dominating current flow along the outer pathway via the NH moiety. When no hydrogen is attached to the inner carbon of the inverted pyrrolic ring, the current prefers the inner route at that ring.
 Please wait while we load your content...
Please wait while we load your content...

