
First principles potential for the cytosine dimer
Abstract
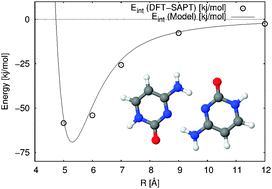
We developed a new first principles potential for the cytosine dimer. The ab initio calculations were performed with a DFT–SAPT combination of the symmetry-adapted perturbation method and density functional theory, and fitted to a model site–site functional form. The model potential was used to predict cluster structures up to cytosine hexamers. The global cluster structure optimizations showed that the new potential is able to reproduce some of the 2D filament structures. Moreover many new non-planar cytosine cluster structures were also discovered. Interaction energies of these clusters were compared with B3LYP-D, MP2, SCS-MP2, SCS-MI-MP2 and AMBER. It has been shown that the model agrees well with all ab initio methods, especially for the cytosine hexamer. The model potential outperforms the AMBER force field and therefore it can be exploited to study more challenging larger systems.
 Please wait while we load your content...
Please wait while we load your content...

