
Crossover among structural motifs in Pd–Au nanoalloys
Abstract
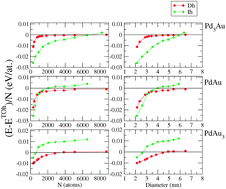
The crossovers among the most abundant structural motifs (icosahedra, decahedra and truncated octahedra) of Pd–Au nanoalloys have been determined theoretically in a size range between 2 and 7 nm and for three compositions equivalent to Pd3Au, PdAu and PdAu3. The chemical ordering and segregation optimisation are performed via Monte Carlo simulations using semi-empirical tight-binding potentials fitted to ab initio calculations. The chemical configurations are then quenched via molecular dynamic simulations in order to compare their energy and characterize the equilibrium structures as a function of the cluster size. For the smaller sizes (of around 300 atoms and fewer) the structures are also optimized at the electronic level within ab initio calculations in order to validate the semi-empirical potential. The predictions of the crossover sizes for the nanoalloys cannot be simply extrapolated from the crossover of the pure nanoparticles but imply stress release phenomena related to the size misfit between the two metals. Indeed, alloying extends the range of stability of the icosahedron beyond that of the pure systems and the energy differences between decahedra and truncated octahedra become asymptotic, around the sizes of 5–6 nm. Nevertheless, such equilibrium results should be modulated regarding kinetic considerations or possible gas adsorption under experimental conditions.
- This article is part of the themed collection: Recent advances in the chemical physics of nanoalloys
 Please wait while we load your content...
Please wait while we load your content...

