
Geometric and electronic structure and magnetic properties of Fe–Au nanoalloys: insights from ab initio calculations
Abstract
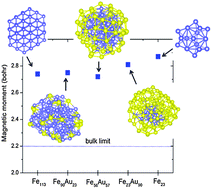
We have performed density functional theory (DFT) based calculations of Fe–Au nanoalloys containing 113 atoms, FexAu113−x (x = 23, 56, 90), to determine their preferred geometric structure and the ensuing electronic structural and magnetic properties. We find that these nanoalloys prefer the formation of a core–shell structure and the Fe core maintains almost a constant magnetic moment of ∼2.8 μB regardless of the Fe content, which is 27% enhancement from the bulk value and in qualitative agreement with some previous results. The local magnetic moment of Fe atoms is well correlated with the local coordination of the Fe atoms. Furthermore, the enhancement of the magnetic moment may be traced to charge depletion from the Fe atoms in the core to the Au atoms in the shell. The preference for the core–shell structure over one with segregated Fe and Au parts could be the low surface tension at the Fe–Au interface, which is larger for the core–shell structure, and can be attributed to strong Fe–Au interfacial interaction as a result of large charge transfer at the interface.
- This article is part of the themed collection: Recent advances in the chemical physics of nanoalloys
 Please wait while we load your content...
Please wait while we load your content...

