
The reactions of N-methylformamide and N,N-dimethylformamide with OH and their photo-oxidation under atmospheric conditions: experimental and theoretical studies†
Abstract
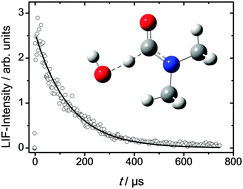
The reactions of OH radicals with CH3NHCHO (N-methylformamide, MF) and (CH3)2NCHO (N,N-dimethylformamide, DMF) have been studied by experimental and computational methods. Rate coefficients were determined as a function of temperature (T = 260–295 K) and pressure (P = 30–600 mbar) by the flash photolysis/laser-induced fluorescence technique. OH radicals were produced by laser flash photolysis of 2,4-pentanedione or tert-butyl hydroperoxide under pseudo-first order conditions in an excess of the corresponding amide. The rate coefficients obtained show negative temperature dependences that can be parameterized as follows: kOH+MF = (1.3 ± 0.4) × 10−12 exp(3.7 kJ mol−1/(RT)) cm3 s−1 and kOH+DMF = (5.5 ± 1.7) × 10−13 exp(6.6 kJ mol−1/(RT)) cm3 s−1. The rate coefficient kOH+MF shows very weak positive pressure dependence whereas kOH+DMF was found to be independent of pressure. The Arrhenius equations given, within their uncertainty, are valid for the entire pressure range of our experiments. Furthermore, MF and DMF smog-chamber photo-oxidation experiments were monitored by proton-transfer-reaction time-of-flight mass spectrometry. Atmospheric MF photo-oxidation results in 65% CH3NCO (methylisocyanate), 16% (CHO)2NH, and NOx-dependent amounts of CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH and CH3NHNO2 as primary products, while DMF photo-oxidation results in around 35% CH3N(CHO)2 as primary product and 65% meta-stable (CH3)2NC(O)OONO2 degrading to NOx-dependent amounts of CH3NCH2 (N-methylmethanimine), (CH3)2NNO (N-nitroso dimethylamine) and (CH3)2NNO2 (N-nitro dimethylamine). The potential for nitramine formation in MF photo-oxidation is comparable to that of methylamine whereas the potential to form nitrosamine and nitramine in DMF photo-oxidation is larger than for dimethylamine. Quantum chemistry supported atmospheric degradation mechanisms for MF and DMF are presented. Rate coefficients and initial branching ratios calculated with statistical rate theory based on molecular data from quantum chemical calculations at the CCSD(T*)-F12a/aug-cc-pVTZ//MP2/aug-cc-pVTZ level of theory show satisfactory agreement with the experimental results. It turned out that adjustment of calculated threshold energies by 0.2 to 8.8 kJ mol−1 lead to agreement between experimental and predicted results.
NH and CH3NHNO2 as primary products, while DMF photo-oxidation results in around 35% CH3N(CHO)2 as primary product and 65% meta-stable (CH3)2NC(O)OONO2 degrading to NOx-dependent amounts of CH3NCH2 (N-methylmethanimine), (CH3)2NNO (N-nitroso dimethylamine) and (CH3)2NNO2 (N-nitro dimethylamine). The potential for nitramine formation in MF photo-oxidation is comparable to that of methylamine whereas the potential to form nitrosamine and nitramine in DMF photo-oxidation is larger than for dimethylamine. Quantum chemistry supported atmospheric degradation mechanisms for MF and DMF are presented. Rate coefficients and initial branching ratios calculated with statistical rate theory based on molecular data from quantum chemical calculations at the CCSD(T*)-F12a/aug-cc-pVTZ//MP2/aug-cc-pVTZ level of theory show satisfactory agreement with the experimental results. It turned out that adjustment of calculated threshold energies by 0.2 to 8.8 kJ mol−1 lead to agreement between experimental and predicted results.
 Please wait while we load your content...
Please wait while we load your content...

