
Dynamics of the A-band ultraviolet photodissociation of methyl iodide and ethyl iodide via velocity-map imaging with ‘universal’ detection
Abstract
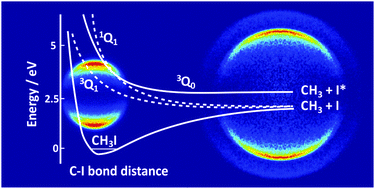
We report data from a comprehensive investigation into the photodissociation dynamics of methyl iodide and ethyl iodide at several wavelengths in the range 236–266 nm, within their respective A-bands. The use of non-resonant single-photon ionization at 118.2 nm allows detection and velocity-map imaging of all fragments, regardless of their vibrotational or electronic state. The resulting photofragment kinetic energy and angular distributions and the quantum yields of ground-state and spin–orbit excited iodine fragments are in good agreement with previous studies employing state-selective detection via REMPI. The data are readily rationalised in terms of three competing dissociation mechanisms. The dominant excitation at all wavelengths studied is via a parallel transition to the 3Q0 state, which either dissociates directly to give an alkyl radical partnered by spin–orbit excited iodine, or undergoes radiationless transfer to the 1Q1 potential surface, where it dissociates to an alkyl radical partnered by iodine in its electronic ground state. Ground state iodine atoms can also be formed by direct dissociation from the 1Q1 or 3Q1 excited states following perpendicular excitation at the shorter and longer wavelength region, respectively, in the current range of interest. The extent of internal excitation of the alkyl fragment varies with dissociation mechanism, and is considerably higher for ethyl fragments from ethyl iodide photolysis than for methyl fragments from methyl iodide photolysis. We discuss the relative advantages and disadvantages of single-photon vacuum-ultraviolet ionization relative to the more widely used REMPI detection schemes, and conclude, in agreement with others, that single-photon ionization is a viable detection method for photofragment imaging studies, particularly when studying large molecules possessing multiple fragmentation channels.
 Please wait while we load your content...
Please wait while we load your content...

