
Modelling analysis of the structure and porosity of covalent triazine-based frameworks†
Abstract
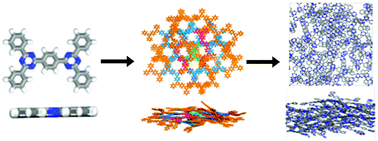
Varying degrees of order have been found experimentally for a series of covalent triazine-based frameworks (CTFs) when synthesised under different reaction conditions. Here, we use molecular modelling to discuss the potential origins of this structural order by analysis of the node and strut building blocks. We use a combination of small model structures based on DFT optimised monomer units and more extended simulations using automated structure growth and molecular dynamics to discuss the influence of the strut structure on the local crystallinity of these materials.
 Please wait while we load your content...
Please wait while we load your content...

